Crouzon
This syndrome was originally described in 1912 by a French neurosurgeon. The syndrome involves fusion or closure of both coronal sutures of the skull and usually occurs due to a specific mutation in a gene (see table below). There is no known cause for this mutation. The incidence can vary but is estimated to be 1:50,000 or more. If a patient chooses to have children there is a 50% incidence of having a child with Crouzon Syndrome.
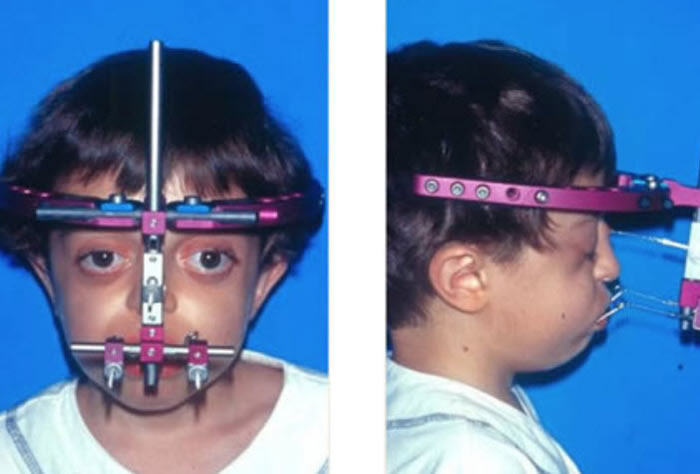
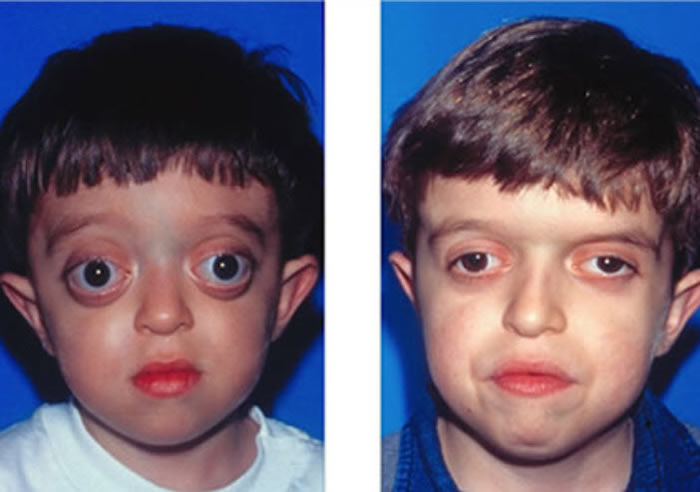
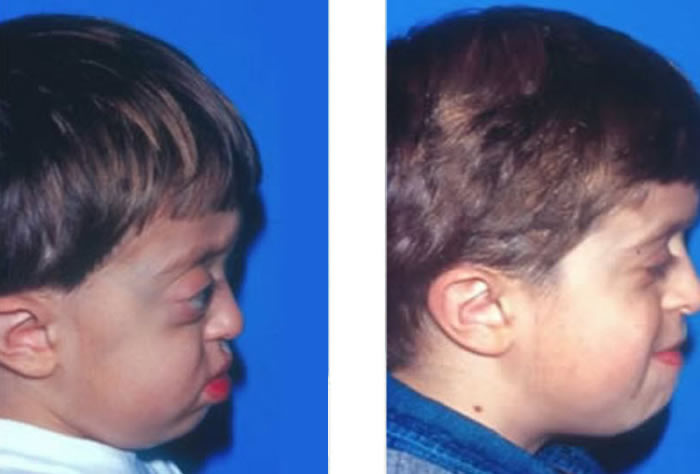
Clinical Features:
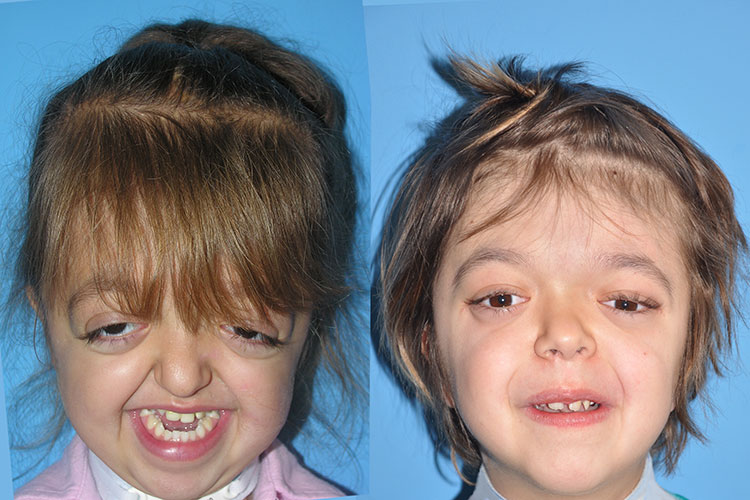
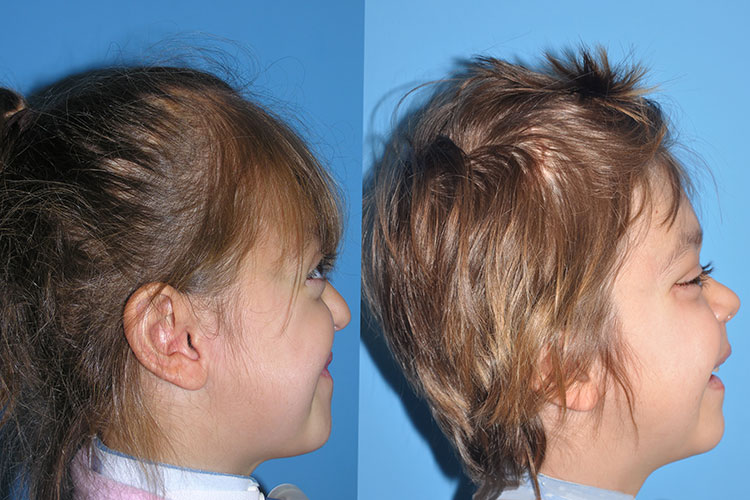
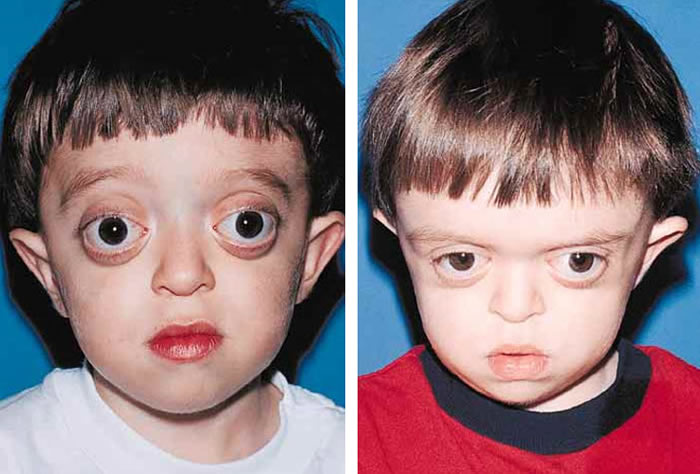
Patients with Crouzon syndrome have very distinct facial features with craniofacial bone morphology similar to Apert syndrome. Premature fusion of the bicoronal suture is the most common type of craniosynostosis with a brachycephalic increase height to the skull. There is retrusion of both the forehead and brow, with midface hypoplasia and shallow orbits with bulging eyes (proptosis). Orbital hpertelorism is less commonly seen in Crouzon’s than Apert’s syndrome. The proptosis is usually more prominent in Crouzon’s than Apert’s; however, in Apert syndrome, the other craniofacial deformities are more complex and severe. The nasomaxillary retrusion may cause some degree of nasal airway obstruction with mouth breathing. The maxillary teeth are retruded resulting in a maloclussion. Other possible abnormalities include conductive hearing loss, inverted V-shape to palate and cornea exposure. Variations of these types of craniofacial problems are seen in the syndromes of Carpenter, Pfeiffer and Saethre-Chotzens.
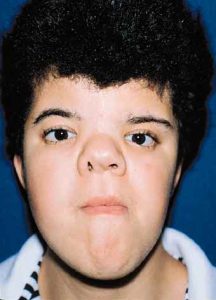
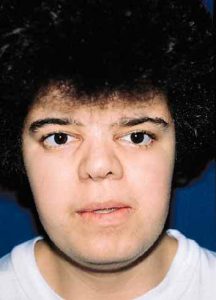
Young woman with Crouzon’s syndrome. Result after correction of hypertelorism, maxillary advancement, mandibular setback and chin reduction.
Treatment:
Our approach to the treatment of Crouzon syndrome is similar to that described for Apert syndrome. The first stage involves treatment of the craniosynostosis with calvarial reshaping/ expansion as needed and frontal-orbital advancement. This procedure is usually performed at 9-12 months and is effective in increasing the intracranial space and enlarging the orbits. The next stage of reconstruction is midface advancement and is usually performed at five to eight years of age. This type of advancement is usually accomplished with a LeFort III osteotomy.The final stage of the reconstruction is management of the dental class III malocclusion. A LeFort I osteotomy may be needed to correct the dental discrepancies in combination with orthodontic intervention. This is usually performed after facial growth is completed and may be combined with a genioplasty (chin reduction/advancement). Additional procedures such as rhinoplasty may be needed.
This is a general description of treatments that may be used. Treatment plans can change and will vary according to the deformity and should be customized for each patient. Discuss the options with your craniofacial surgeon.