Apert Syndrome
Apert syndrome or acrocephalosyndactyly syndromes are rare conditions. In 1906, Apert described the skull, facial, and hand deformities of several patients characteristic of this syndrome that now bears his name. The incidence of infants born with Apert syndrome is one for every 100,000 to 160,000 live births. Many of the infants born with this syndrome show a sporadic transmission, which means that a family may have a child with Apert’s when no other members of the family are affected. The recurrent risk of having another child with Apert’s for two unaffected parents is negligible. However, if the parent is affected there is a 50% chance of each offspring having Apert syndrome with both males and females affected equally.
Clinical Features:
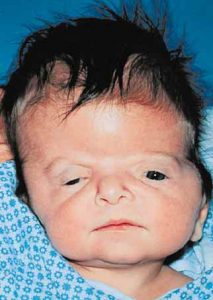
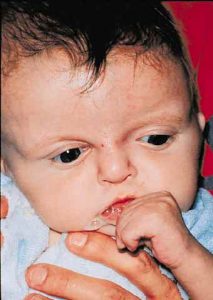
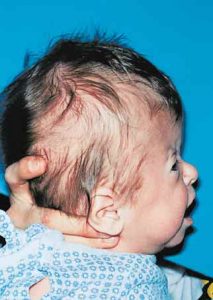
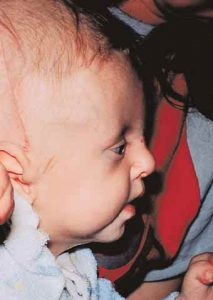
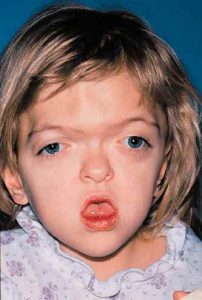
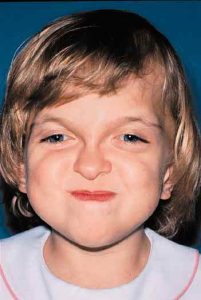
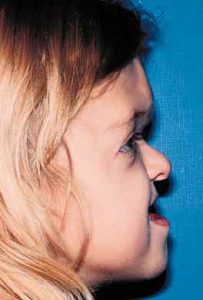
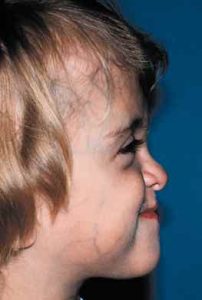
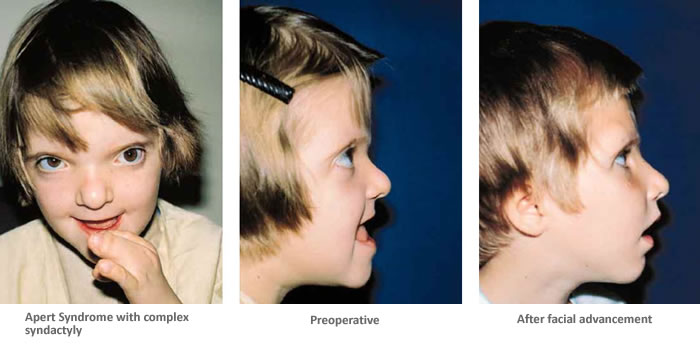
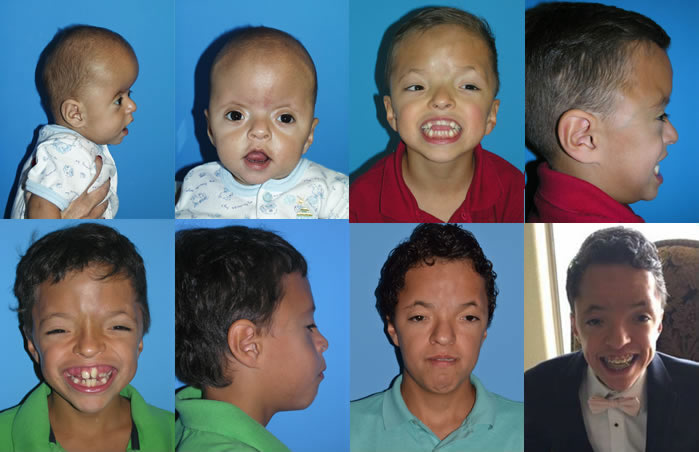
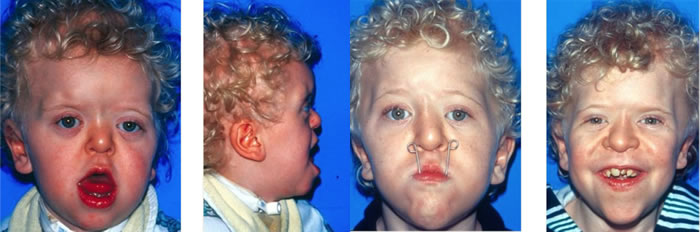
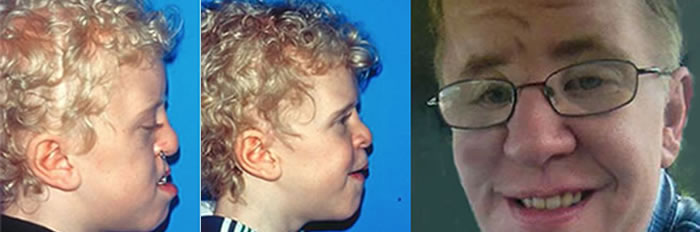
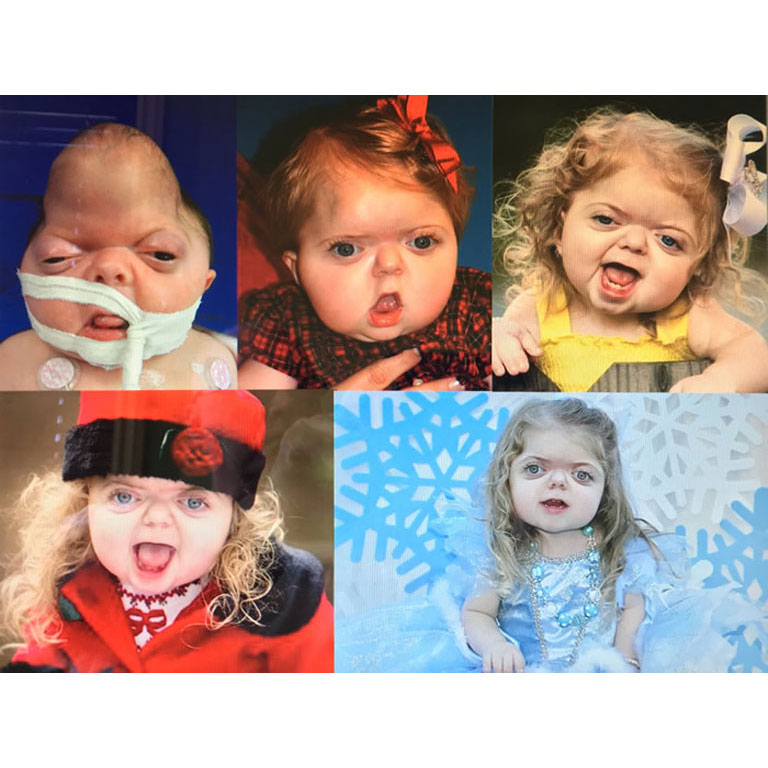
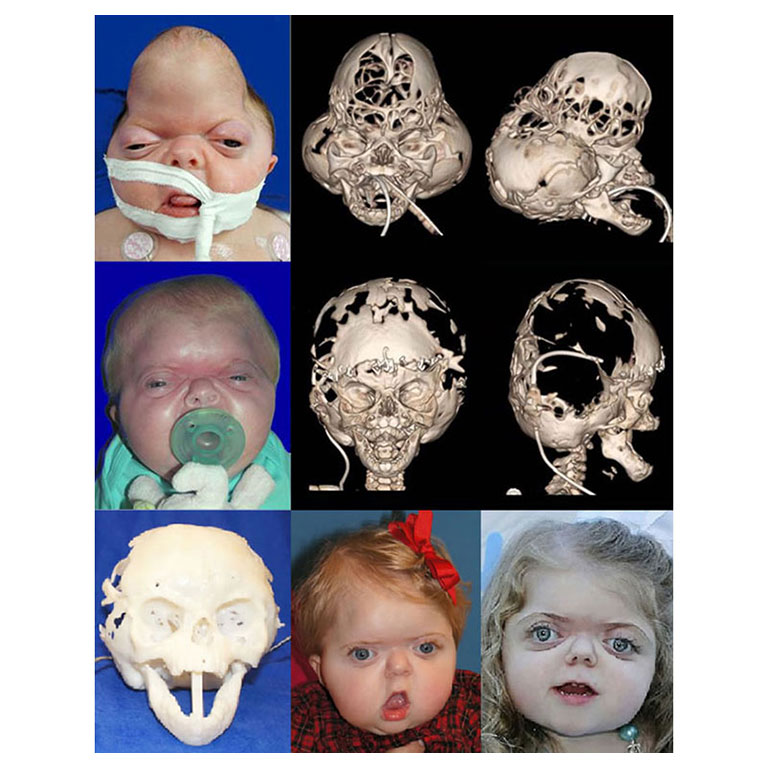
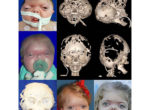
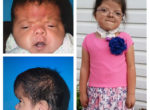
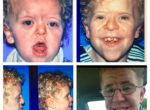
Patients with Apert syndrome have very distinct facial and extremity features. Abnormal skull shape is due to craniosynostosis or premature fusion of the sutures or soft spots. The skull usually demonstrates a short anteroposterior diameter (brachycephaly) and may be excessively tall (turricephalic) or abnormally wide (euryprosopia). The forehead is generally always retruded, but this may not be obvious due to the hypoplasia of the midface. The orbits or bony sockets which contain the eyes are very shallow causing a bulging or proptosis. The orbits are usually rotated downward and lateral causing a downward shape to the lateral corners of the eyes. There may be a moderate increased distance between the eyes (hypertelorism) with muscle imbalance.
The middle of the face in Apert’s is both retruded and very hypoplastic. This causes the central midface to have a characteristic sunken in appearance with the nose being thick and beaked. The upper jaw or maxilla characteristically shows a narrow arch with an open bite and dental crowding. The maxilla is significantly retruded compared to the mandible with the teeth of the lower jaw projecting in front of the upper teeth. Other possible clinical features include moderate hearing loss, speech impairment, acne, and decreased mental capability in some individuals. Intellectual potential may be difficult to evaluate due to communication problems. Some patients with Apert syndrome may have normal intelligence. All patients with Apert syndrome demonstrate a unique hand malformation. This is characterized by a complex syndactyly or fusion of the skin, soft tissue, and bones of the fingers. Both hands are affected equally, as are the feet. This unusual variation of syndactyly can be used to identify Apert’s from other similar syndromes.
Treatment:
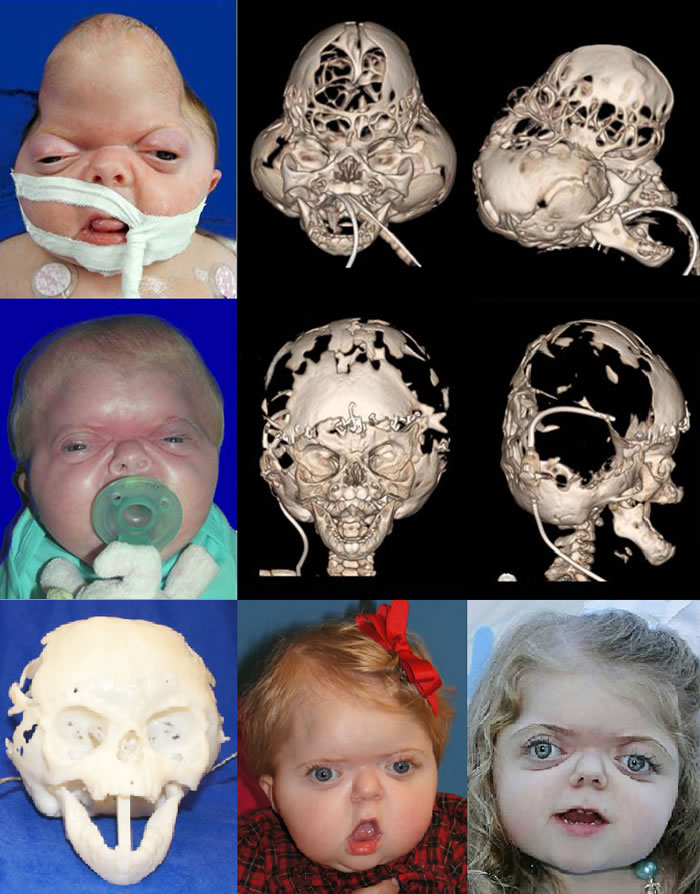
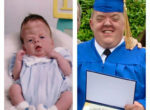
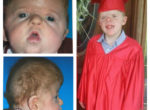
The treatment of patients with Apert syndrome is not uniform due to significant variations in the facial anomalies, age of patients when first seen, and previous operations. Our primary concern of the infant born with this syndrome is: compression of the brain, breathing problems, protruding eyes with corneal exposure, and lack of facial growth. The surgical plan must be flexible and individualized to the patient. Multiple stages or operations at different ages are usually necessary.
When we see these patients as infants the first step is to have a multi-disciplined evaluation to assess the severity of the problems to organize a coordinated comprehensive treatment plan. Giving the brain enough room for growth with release of skull fusion is a priority and may require posterior and anterior skull distraction performed in stages. Reshaping and moving the forehead and brow forward is important to protect the globes. A midface advancement or distraction is needed to correct the maxillary hypoplasia. A variety of options are now available to correct these complex problems. Each patient should have a customized surgical plan that meets their needs. You should discuss the options with your craniofacial surgeon.
The final steps to the reconstruction that may be needed are maxillary/mandibular osteotomies to complete the correction of any further dental discrepancies. These procedures are usually performed after eruption of permanent dentition and completion of growth (teen years). Additional procedures such as rhinoplasty, genioplasty and eyelid surgery may be beneficial.
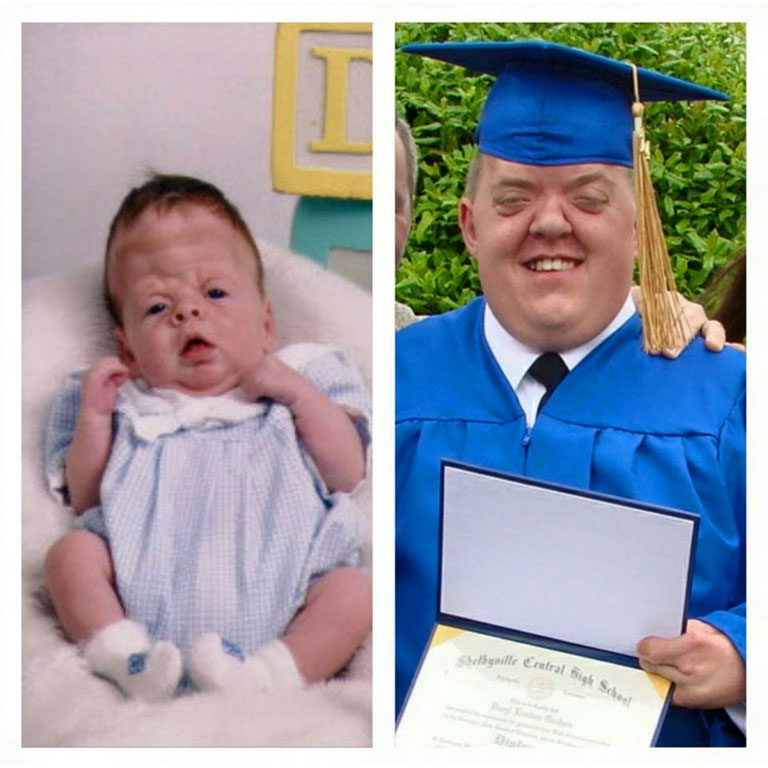

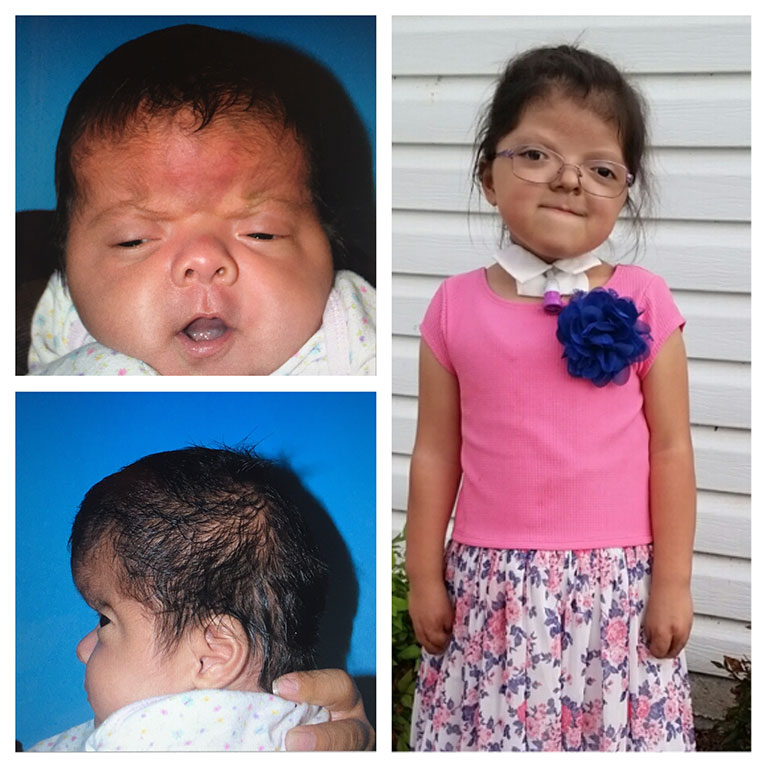
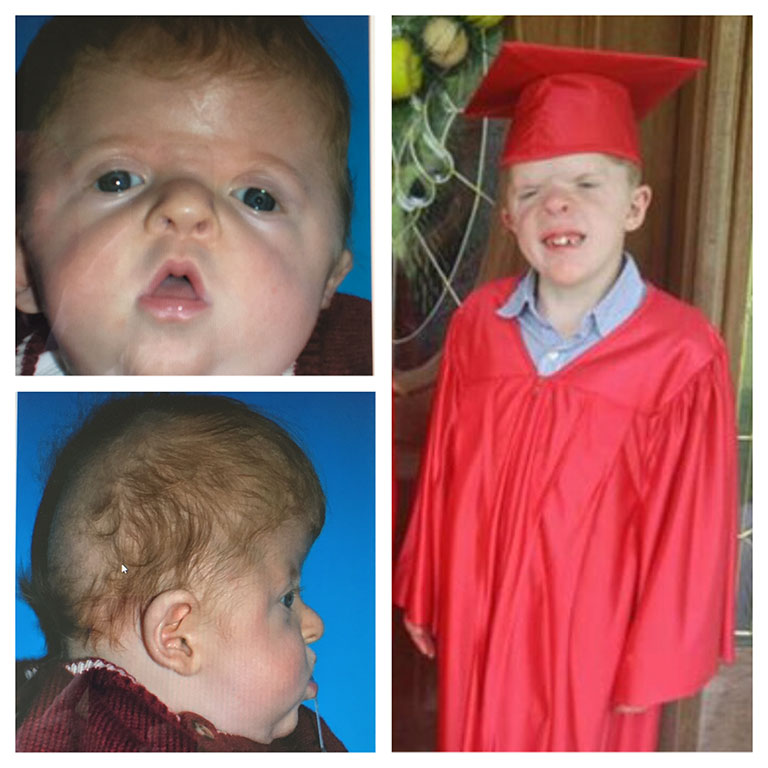
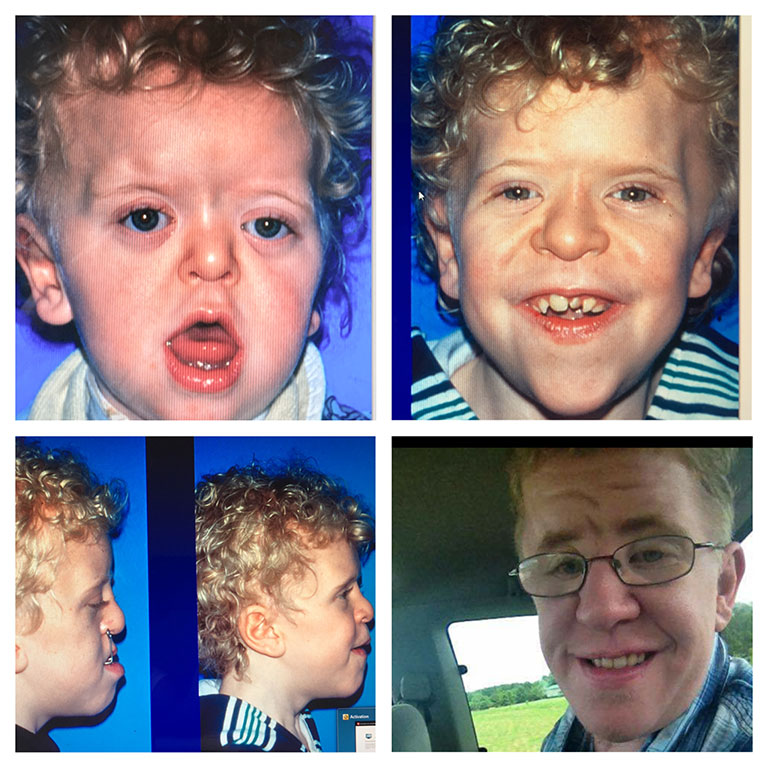


Click here to read of the Use of Osteogenesis Distractors in Cloverleaf Skull Reconstruction by Larry A. Sargent, MD and Devin Griner, MD