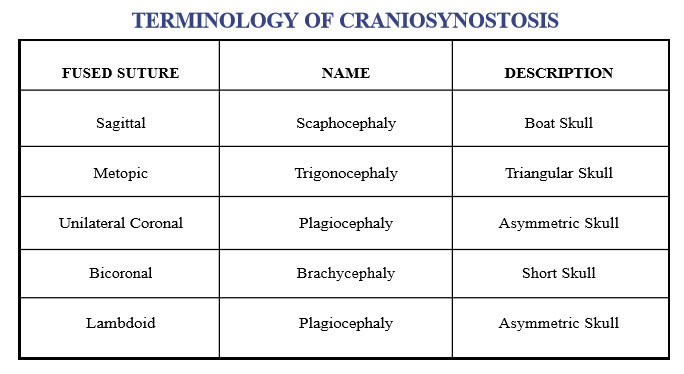
Craniosynostosis
Craniosynostosis can be defined as the premature closing of one or more of the normally present bony gaps between the different bones of the skull. These linear areas where the bones have not yet healed together are called cranial sutures. They intersect in certain areas of the skull forming “soft spots” or fontanelles. The cranial suture lines or non-ossified zones accommodate the brain’s growth and expansion during the first years of life. The brain increases in volume about 2-1/2 to 3 times the first two years of life with these nonossified areas of bone remaining open for a variable length of time and then closing in a predictable manner. Premature synostosis or closure of the suture will stop the growth of the skull in the direction perpendicular to that suture; however, growth will continue in the direction parallel to the suture. The result is that synostosis of a particular suture will alter the shape of the skull in a predictable manner with recognizable patterns. With multiple suture involvement, increased intracranial pressure can occur since the skull cannot sufficiently expand to accommodate normal brain growth. In moderate craniosynostosis with only one suture involved increased intracranial pressure has been found to be present in approximately 13-23% of patients.
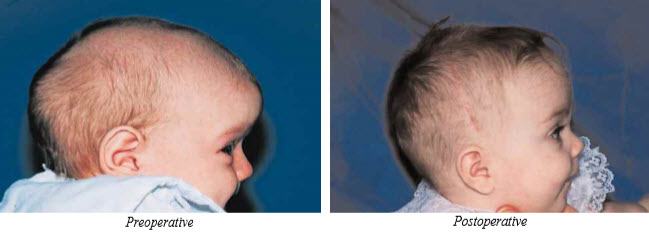
The surgical treatment of craniosynostosis has been transformed by the development and applications of craniofacial techniques to reshape the skull and upper face in the infant. Functionally, the goal in treatment is to release the fused sutures to prevent any problems associated with increased intracranial pressure as well as creating the potential for normal growth. Cosmetically, the goal is to obtain normal shape of the skull and face, thus minimizing psychosocial problems.
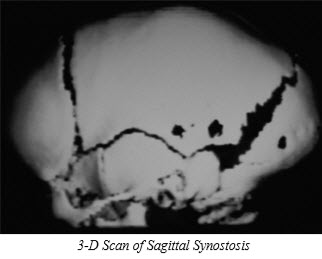
Approximately one infant in every one thousand births will have a premature closure of a cranial suture associated with a skull deformity. Infants born with abnormal skull shapes should be suspected of having craniosynostosis. However, abnormal skull shapes at birth may be related to fetal head position or birth trauma; this type of deformity will usually correct itself in several months. True craniosynostosis will not improve with time and usually worsens with growth. On examination palpation of the abnormal portion of the head often reveals a ridge in the area of the fused suture. Once craniosynostosis is suspected, the diagnosis can be verified with plain skull radiographs and CT scans. We routinely obtain CT scans with three-dimensional reconstructions to confirm the diagnosis, rule out other intracranial abnormalities, and to fully assess the deformity. The 3-D scans can very clearly show the skull shape and help in pre-operative planning. Isolated craniosynostosis must be distinguished from a syndrome that has as one of its components craniosynostosis. The specific diagnosis is an important starting point in treatment planning.
Etiology:
While the pattern of embryonic craniofacial development has been well defined through extensive research, very little is known about the etiology of many craniofacial anomalies. There is strong evidence for a genetic role in the occurrence of some craniosynostosis. An inherited tendency or family history has been reported in many cases in the literature, however, most cases of isolated craniosynostosis are usually sporadic in occurrence with no known etiology.
Team Evaluation:
Patients with craniosynostosis need to be evaluated and treated at a recognized craniofacial center with a team approach. We have a multidisciplinary team at our center to provide the comprehensive care necessary to adequately evaluate and treat craniofacial patients and their families. Members of the craniofacial team work together to insure that the patients are evaluated and treated in a coordinated manner and that all their needs, both physical and psychosocial, are met. The team combines the expertise of each specialist to provide a level of comprehensive care that cannot be provided by a single physician. The primary physician or pediatrician plays a key role in early diagnosis and referral so that a successful treatment is accomplished.
Surgical Treatment:
Tremendous advancements have been made in the treatment of infants with isolated craniosynostosis due to the application of craniofacial surgery principles. Past treatments such as strip craniectomies which remove only the fused suture have resulted in unpredictable results. These procedures have often resulted in skull and facial deformities that are difficult to correct in the older child. Current surgical treatment involves a team approach with a combined expertise of a neurosurgeon and craniofacial surgeon. The goal of surgery is to establish normal contour of the brow, forehead and skull; and to allow the brain to grow and expand in a normal manner with release of synostosis. The timing of surgical correction depends on the age at diagnosis and referral. In our experience we feel that the best results are obtained when infants undergo surgical repair between 9-12 months of age. This period has several advantages:
- remodeling is easier because the bone is very malleable;
- rapid brain growth benefits bone remodeling;
- bone defects heal rapidly.
Early correction also prevents further deformity caused by unreleased, fused sutures. Therefore, early diagnosis and referral to a craniofacial center is crucial. We advocate extensive release of the fused suture, total reshaping of the brow, superior orbits, and skull as necessary. Older patients will require modification of this plan due to the presence of more rigid bone and slower cranial growth. In addition, older patients may require reconstruction of secondary facial deformities.
In the event that intraoperative or postoperative blood transfusions are needed, the parents are asked to provide designated donor blood. In addition, aspirin and other medications that might prolong the blood clotting time are to be avoided.